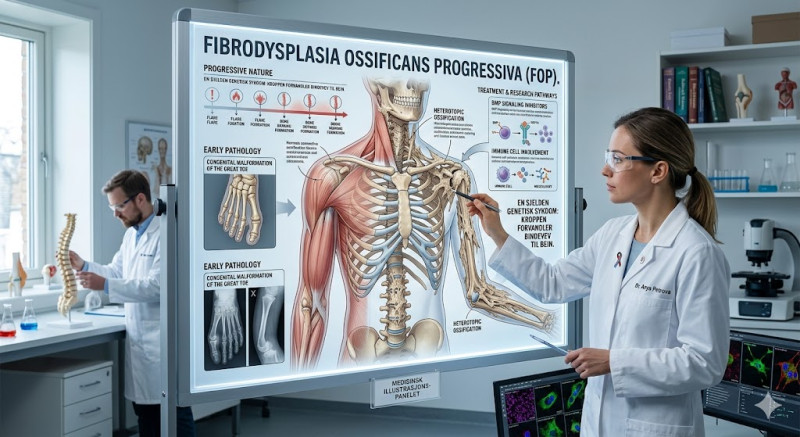
Fibrodysplasia Ossificans Progressiva (FOP), historisk kjent som "Myositis Ossificans Progressiva", er en ekstremt sjelden og ødeleggende genetisk bindevevssykdom. Den rammer omtrent én av to millioner mennesker på verdensbasis. Sykdommen er preget av at kroppens reparasjonsmekanisme svikter fundamentalt: bindevev, sener, ligamenter og skjelettmuskulatur blir gradvis omdannet til ekte beinvev. Denne prosessen kalles heterotopisk ossifikasjon. Over tid dannes det et "ekstra skjelett" på utsiden av det normale skjelettet, noe som gradvis låser kroppens ledd og fører til permanent immobilisering.
Sykdomsforløp og symptomer
Det første kliniske tegnet på FOP er nesten alltid en medfødt misdannelse av stortærne, som er kortere enn normalt og bøyd innover. I løpet av barndommen, vanligvis før tiårsalderen, begynner de første episodene med smertefulle hevelser i bløtvevet, ofte utløst av mindre traumer som fall, slag eller intramuskulære vaksiner. Disse hevelsene (såkalte flare-ups) forsvinner etter hvert, men etterlater seg nydannet beinvev. Prosessen starter vanligvis i nakken og skuldrene, og beveger seg gradvis nedover ryggen og ut i ekstremitetene. Når leddene blir innkapslet i bein, mister pasienten evnen til å bevege armer, bein og til slutt kjeven, noe som gjør spising og tale ekstremt vanskelig.
Diagnose og forskning
Diagnostisering av FOP er kritisk, fordi feildiagnostisering (ofte som kreft eller dype vevsinfeksjoner) kan føre til at leger utfører biopsier. Kirurgiske inngrep eller vevsprøver er katastrofale for FOP-pasienter, da ethvert fysisk traume akselererer beindannelsen dramatisk. Sykdommen skyldes en mutasjon i ACVR1-genet, som koder for en reseptor i kroppens beinmorfogenetiske proteinsystem (BMP). Det finnes i dag ingen kur som kan reversere prosessen, og kirurgisk fjerning av det ekstra beinet fører bare til at det vokser tilbake enda mer aggressivt. Behandlingen er derfor støttende, med fokus på smertestilling og forebygging av traumer.