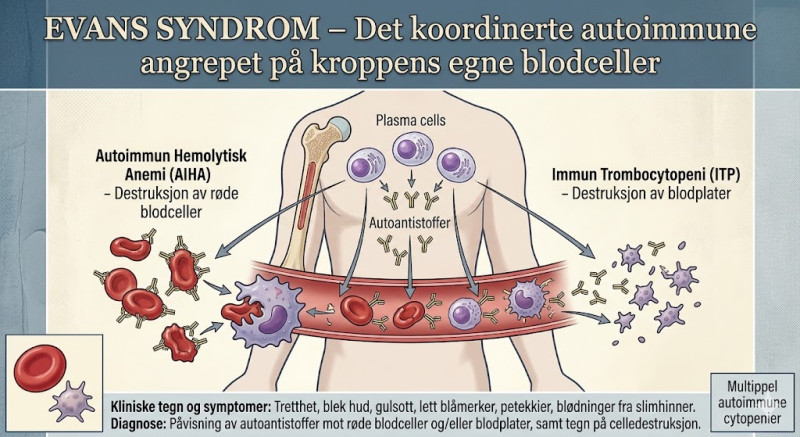
Evans syndrom er en sjelden og potensielt livstruende autoimmun tilstand som representerer et alvorlig sammenbrudd i immunsystemets evne til selvtoleranse i det hematologiske (blodrelaterte) systemet. Sykdommen er unikk fordi den ikke retter seg mot ett spesifikt organ eller én enkelt celletype, men kjennetegnes av at kroppen produserer autoantistoffer som angriper og ødelegger to eller flere forskjellige typer blodceller samtidig, eller etter hverandre i tid. Evans syndrom defineres formelt som en kombinasjon av to separate autoimmune tilstander: autoimmun hemolitisk anemi (AIHA) og immun trombocytopeni (ITP), og i sjeldne tilfeller også autoimmun nøytropeni.
For å forstå alvorlighetsgraden av Evans syndrom, må man se på funksjonen til de cellene som blir utryddet. Ved autoimmun hemolitisk anemi produserer pasientens B-lymfocytter antistoffer (vanligvis av typen IgG) som binder seg til overflaten på de røde blodcellene (erytrocyttene). Når disse merkede blodcellene passerer gjennom milten og leveren, gjenkjenner makrofager (eteceller) antistoffene og destruerer de røde blodcellene i høyt tempo (hemolyse). Siden de røde blodcellene transporterer oksygen til kroppens vev, fører dette til en akutt og alvorlig anemi. Samtidig, ved immun trombocytopeni, retter en annen populasjon av antistoffer seg mot blodplatene (trombocyttene), som er ansvarlige for at blodet koagulerer ved skader. Blodplatene ødelegges på samme måte i milten, noe som fører til en kritisk mangel på koagulasjonsevne.
Det kliniske bildet ved Evans syndrom er preget av de kombinerte effektene av cellemangelen, og symptomene kan svinge voldsomt og uforutsigbart. På grunn av anemien opplever pasienten en ekstrem og lammende utmattelse, blekhet, svimmelhet, hjertebank og kortpustethet ved de minste anstrengelser. Nedbrytningen av de røde blodcellene frigjør store mengder bilirubin, noe som fører til at pasienten utvikler gulsott (gulfarging av hud og øyne) og mørk urin. På grunn av mangelen på blodplater (trombocytopeni) utvikler pasienten en uttalt blødningstendens. Dette viser seg som spontane blåflekker (ekchymoser), små punktformede blødninger i huden (petekkier), hyppige neseblødninger, blødende tannkjøtt, og i verste fall livstruende indre blødninger i hjernen eller fordøyelseskanalen. Hvis pasienten i tillegg har nøytropeni (mangel på hvite blodceller), blir kroppen ekstremt sårbar for alvorlige bakterielle infeksjoner.
Diagnosen stilles gjennom omfattende blodanalyser. En direkte Antiglobulintest (Coombs test) brukes for detektere antistoffene som sitter fast på de røde blodcellene. En fullstendig blodstatus vil vise markant lave verdier av både hemoglobin og trombocytter, mens benmargsbiopsi gjøres for å utelukke underliggende leukemi eller lymfekreft, da Evans syndrom ofte kan oppstå sekundært til andre lymfoproliferative sykdommer eller revmatiske lidelser som Lupus (SLE). Behandlingen er vanskelig fordi sykdommen er beryktet for å være refraktær – det vil si motstandsdyktig mot behandling – og den har en høy tendens til tilbakefall. Første linje er høye doser med kortikosteroider for å bremse antistoffproduksjonen. Hvis dette svikter, må man ty til biologiske medisiner som rituximab (et monoklonalt antistoff som utrydder de B-cellene som lager antistoffene), immundempende midler som cyklosporin, eller kirurgisk fjerning av milten (splenektomi) for å stoppe det fysiske stedet hvor blodcellene blir ødelagt.